Mit Alignment steht den Foldit-Spielern seit Februar 2010 nun auch ein Hauptwerkzeug professioneller Proteinstrukturvorhersage zur Verfügung. Dabei wird bei Proteinen mit unbekannter Tertiärstruktur geschaut, ob ähnliche Aminosäuresequenzen auch bei Proteinen mit bereits bekannter Struktur zu finden sind. Wird man fündig, dann versucht man, Strukturen bekannter Sequenzen auf das "neue" Protein zu übertragen.
Funktionsweise bei Foldit[]
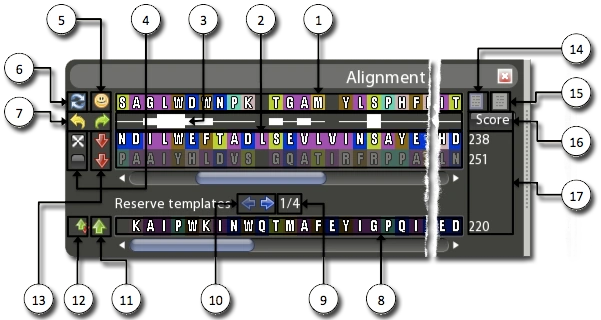
In der obersten Reihe sehen wir die Sequenz "unseres" Proteins (1). Die Buchstaben stehen für die entsprechenden Aminosäuren lt. Einbuchstabencode. In den weiteren Reihen stehen Sequenzen mit bekannter Struktur (2). Wie gut diese mit "unserer" Sequenz übereinstimmen, kann man in dem Bereich zwischen der ersten und der zweiten Reihe sehen. Je dicker der weiße Balken, desto größer die Übereinstimmung an dieser Stelle (3). Am größten ist sie natürlich bei identischen Aminosäuren (L/L, W/W, S/S, usw.) aber auch ähnliche Aminosäuren - zu erkennen an gleichen Farben (G/I, D/E, T/S, usw.) - besitzen noch eine gewisse Übereinstimmung.
Jeweils rechts von den Sequenzen steht ein Gesamtpunktwert für die jeweilige Übereinstimmung mit der Sequenz "unseres" Proteins (17). Je höher dieser Wert, desto besser.
Innerhalb jeder Sequenz können alle Aminosäuren nach links und rechts verschoben werden, wobei ihre Reihenfolge natürlich nicht verändert werden kann. Die veränderten Übereinstimmungen schlagen sich sofort in der Dicke des weißen Balkens und der Höhe des Scores nieder.
Funktion der Buttons[]
- Die beiden blauen Pfeile (6) in der linken Schaltfläche setzen das angekreuzte Alignment in seinen Ausgangszustand zurück. Ist keines angekreuzt, werden alle Alignments und auch die Sequenz "unseres" Proteins zurückgesetzt.
- Der gelbe Smiley (5) versucht, das angekreuzte Alignment auf "unser" Protein zu übertragen. (Häufig funktioniert das auch nicht.) Ist kein Alignment angekreuzt, wird das oberste genommen.
- Der gelbe Pfeil nach links (7) macht die letzte Änderung rückgängig (Undo).
- Der grüne Pfeil nach rechts (7) macht das letzte Undo rückgängig (Redo).
- Das angekreuzte Alignment (4) ist das gerade aktive, das auch die Proteinansicht überlagert.
- Der grüne Pfeil nach oben (11) verschiebt das entsprechende Alignment an die Spitze.
Proteinansicht bei Alignments[]

{kind=link}
Abb.2: Markiertes Segment des Proteins und seine Entsprechung im Modell

{kind=link}
Abb.3: Markierte Segmente des Alignments und ihre Entsprechungen im Modell
Das jeweils aktive Alignment wird in der Proteinansicht gemeinsam mit der Sequenz "unseres" Proteins in einem farbigen Modell visualisiert, dass die transparent angezeigte aktuelle Struktur des Proteins überlagert. Die Segmente des Alignments entsprechen dabei den identisch eingefärbten Kugeln im Modell, die Segmente "unseres" Proteins entsprechen den identisch eingefärbten Röhren im Modell.
Befindet sich in der Sequenz des Alignments ein Gap (eine Lücke), dann fehlen im Modell die Kugeln zwischen den Röhren. Befindet sich der Gap hingegen in der Sequenz "unseres" Proteins (Abb.2 zwischen E und W), dann sind im Modell einige Kugeln nur mit dünnen Strichen verbunden (dünner Pfeil in Abb.3). Nach einem Threading (der Übertragung eines Alignments auf das Protein), werden durch Gaps so einige Segmente entweder unnatürlich gestaucht oder gestreckt. Diese Segmente haben dann Punktwerte im extremen Minusbereich und müssen später mit Rebuilds oder lokalen Wiggles bearbeitet werden.
Bemerkungen[]
Für die Arbeit mit dem Alignment-Werkzeug empfiehlt es sich, unter den Ansichtsoptionen Show outlines ein- und Dark background auszuschalten. So kann man die aktuelle Proteinstruktur, die ja nur transparent angezeigt wird, am besten sehen.
Nach dem Threading sollte zuerst Shake und dann Wiggle Sidechains auf das gesamte Protein angewendet werden. Danach kann man Teile des Proteins, soweit gewünscht, mit Gummibändern stabilisieren, bevor man einen globalen Wiggle startet.
Es ist sehr wahrscheinlich, dass der Score des Puzzles danach immer noch auf Null steht. Deshalb ist es sinnvoll, vorher schon unter Menu --> General Options "Show advanced GUI" zu aktivieren. Der Score wird dann ggf. auch als Negativwert angezeigt. Außerdem sollte in den Ansichtsoptionen Use relative score coloring aktiviert sein. So ist es einfacher, die jeweils schlechtesten Segmente zu finden und gezielt zu bearbeiten. (Um sich den Punktwert und weitere Informationen zu einem einzelnen Segment anzeigen zu lassen, kann man die Tab(ulator)-Taste drücken, während sich der Mauszeiger über diesem Segment befindet.)
Das Alignment-Werkzeug kann nur über das Standardinterface (über Actions --> Show Alignment) aufgerufen werden. Im Auswahl-Interface geht das (noch) nicht.